
10 Minute Standard Presentations 2024
Session 1 – Wednesday kl 15.00-16.00

Agnes Beenfeldt Petersen
Agnes B. Petersen1, Anita Solem1, Gerd Inger Sætrom1, Håvard Sletta2, Finn L. Aachmann1, Anne Tøndervik2,1, Jochen Schmid2, & Gaston Courtade1
(1) Department of Biotechnology and Food Science, Norwegian University of Science and Technology. Trondheim, Norway, (2) Department of Biotechnology and Nanomedicine, SINTEF Industry. Trondheim, Norway
Alginate epimerases convert β-D-mannuronate (M) to its C-5 epimer α-L-guluronate (G) in alginates. Alginates are linear anionic polysaccharides produced by brown seaweed and some bacteria. In alginates M and G are organized in blocks of M (polyM), blocks of G (polyG), and in alternating blocks of MG (polyMG). [1]
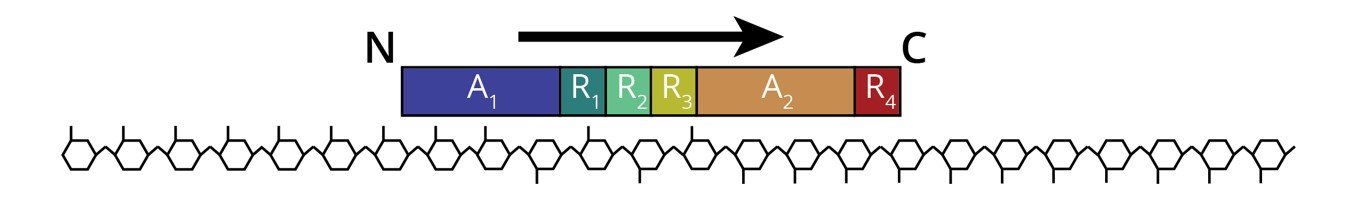
Alginate epimerases are modular, Ca2+-dependent enzymes that work in a processive manner [1] The alginate epimerase AlgE1 from Azotobacter vinelandii consists of two catalytically active A-modules (A1 and A2) and four carbohydrate binding R-modules (R1-R4) (Figure 1) [2]. Previous studies show that A1 introduces G to both polyMG and polyM, while A2 creates MG-blocks from polyM, when the A-modules and their subsequent R-modules are expressed separately [2]. The present study seeks to better understand how the modules function together, and how they influence substrate binding and enzyme processivity. The main methods used were mutational studies combined with NMR spectroscopy.
This study consists of three parts. In the first part of the study, the two A-modules were inactivated, which confirmed the specificity of the A-modules. To enable mutation, a small three amino acid residue motif was inserted in the beginning of A2, which disrupted their processiveness and substrate binding.
The second part of the study investigated what happens when the relative position of A1 and A2 are interchanged, and when A2R4 are switched to the N-terminal of the enzyme. Both modifications changed the mode of action of AlgE1.
Lastly, the A-modules of AlgE1 were replaced with the A-module of AlgE7, which is similar in structure, but performs both epimerisation and lyase reactions. This showed that AlgE1 processes along the alginate chain with the C-terminal end first (see Figure 1).
Overall, the study widens the understanding of the role of the two catalytic modules of AlgE1 and how they function together.
References:
- Petersen AB, Tøndervik A, Gaardløs M, Ertesvåg H, Sletta H, and Aachmann FL (2023) Mannuronate C-5 Epimerases and Their Use in Alginate Modification. Essays in Biochemistry, 67(3): 615-627
- Ertesvåg H, Høidal HK, Skjåk-Bræk G, and Valla S (1998) The Azotobacter vinelandii Mannuronan C-5-Epimerase AlgE1 Consists of Two Separate Catalytic Domains. Journal of Biological Chemistry, 273(47) 30927-30932

Greta Daae Sandsdalen
Greta Daae SandsdalenI1, Maryam Imam2, Ole Morten Seternes3, Adele Williamson4 & Hanna-Kirsti Schrøder Leiros1
(1) Biomolecular and Structural Chemistry, Department of Chemistry, UiT The Arctic University of Norway, (2) The Norwegian College of Fishery Science, UiT The Arctic University of Norway, (3) Department of Phamacy, UiT The Arctic University of Norway, (4) The University of Waikato, New Zealand
The CRISPR-Cas genome editing system has revolutionized molecular biology, providing an array of biotechnological tools for carrying out precision genome modification and regulation. One limitation of the system at present is that most available tools are developed from and optimized for mesophilic organisms, which limits their utility in cold living organisms. This paucity of available knowledge on psychrophilic CRISPR-Cas systems and affiliated genome editing tools is particularly problematic for researchers of cold-blooded eukaryotes, where mismatched thermal preferences of the CRISPR components hinder application efficiency.
This project is one of three interdisciplinary components of the UiT Strategic-Funded ‘FISH&CRISPR Innovative strategies to improve salmon health’ which aims to establish a platform for the development of a low-temperature CRISPR-Cas genome editing system optimized for salmonids.
In this project, the main goal is to discover and develop one or more CRISPR-associated endonucleases for efficient and precise genome editing at low temperatures. A second aim is to gain insights on CRISPR-systems across cold-adapted bacteria through bioinformatics analysis.
Our findings reveal a low prevalence of CRISPR-Cas systems in cold-adapted bacteria, compared to mesophilic and thermophilic species, where only 17.7% of the analyzed genomes contained CRISPR operons. Further, five CRISPR endonucleases were selected for experimental characterization. Currently, one of them shows promise for genome editing applications in low-temperature conditions.
The identification and initial characterization of Cas endonucleases from cold-adapted bacteria mark a pivotal step towards establishing a CRISPR-Cas platform optimized for salmonids. This advancement could significantly impact genomic studies and biotechnological applications for cold-adapted organisms, aligning with the goals of enhancing salmon health and aquaculture sustainability.

Hemanga Gogoi
Hemanga Gogoi, Dario Segura-Peña, & Nikolina Sekulic
Centre for Molecular Medicine Norway, University of Oslo
The chromosomal passenger complex (CPC) is a macromolecular assembly comprising four subunits: Aurora B kinase, Inner Centromere Protein (INCENP), Survivin, and Borealin. This complex plays a role in the regulation of chromosomal segregation during mitosis. Additionally, Shugoshin 1 (Sgo1) ensures chromosome stability as it protects the cohesin, through recruitment of two protein complexes with opposite enzymatic activities, the Protein phosphatase 2A (PP2A) and the CPC. The enzymatic component of the CPC is the serine/threonine protein kinase Aurora B. Localization of Aurora B to the inner centromere is critical for proper chromosome segregation and therefore chromosome stability. Sgo1, plays an important role in the recruitment of the CPC to the inner centromere but the biochemical and structural details of the Sgo-CPC interaction are not understood. Here we undertake a biochemical and structural approach to understand this critical interaction, base in Hydrogen deuterium exchange, mutagenesis, and Fida-Bio experiments, we found that borealin is the subunit of the CPC that is more engaged in interaction with Sgo1. In addition, we show in-vitro that both the CPC and PP2A can interact simultaneously with Sgo1 and propose a model for the formation of this ternary complex.

Jan Benedict Spannenkrebs
Jan Benedict Spannenkrebs, Agnes Beenfeldt Petersen, & Prof. Johannes Kabisch
NTNU, Institute for Biotechnology and Food Science
Bacillus subtilis can form metabolically dormant endospores in response to deteriorating environmental conditions such as poor nutrient availability. In a process called sporulation, a Bacillus subtilis cell divides asymmetrically. In one half of the cell a copy of the DNA is enclosed in multiple membrane and proteinaceous layers. Once sporulation concludes, the mother cell lyses, releasing the metabolically dormant spore into the environment. The formed spore provides protection for the DNA against extreme / harmful conditions, including high temperatures, desiccation, radiation and irritating chemicals. The spore core, which contains the DNA, is encased in an inner and outer coat. The outermost layer, designated the “crust,” is primarily composed of six proteins (CotV, W, X, Y, Z, and CgeA)[1].
These crust proteins as well as others from different spore layers have already been used to create fusion proteins with proteins of interest, for example enzymes or antigens. For this, a copy of the fusion proteins DNA is inserted into B. subtilis under the control of a sporulation specific promoter. These fusion proteins self-assemble into the respective spore layer during sporulation. This system has for example been used for the display of a photodecarboxylase for the transformation of lipids to hydrocarbons[2], as well as for the display of the receptor binding domain of SARS-CoV-2[3]. The immobilization of proteins in this manner greatly facilitates downstream purification. Due to the spores’ size, purification is possible by comparatively low-tech means like repeated centrifugation and washing.
A novel candidate class of enzymes for spore display are Alginate epimerases such as the processive AlgE-type. Alginates are linear polysaccharides comprised of linked β-D-mannuronate (M) and its C-5 epimer α-L-guluronate (G). The G-blocks can chelate divalent cations like Ca2+, resulting in a hydrogel. The properties of the hydrogel can be modified by altering the ratio and sequence of the M and G blocks, making them an interesting product for example the food and pharmaceutical industry[4]. Alginate epimerases are enzymes capable of epimerizing M into G blocks, thus modifying the alginates properties.
Join this talk to get an insight into recent developments in spore displayed enzymes in our lab, including the display of epimerases and how we can measure their activity in real-time.
References:
- Bartels, J., Blüher, A., López Castellanos, S., Richter, M., Günther, M. and Mascher, T. (2019), The Bacillus subtilis endospore crust: protein interaction network, architecture and glycosylation state of a potential glycoprotein layer. Mol Microbiol, 112: 1576-1592
- Karava, M.; Gockel, P.; Kabisch, J. Bacillus Subtilis Spore Surface Display of Photodecarboxylase for the Transformation of Lipids to Hydrocarbons. Sustainable Energy Fuels 2021, 5 (6), 1727–1733.
- A. Vetráková, R. Kalianková Chovanová, R. Rechtoríková, D. Krajčíková, I. Barák, Bacillus subtilis spores displaying RBD domain of SARS-CoV-2 spike protein, Computational and Structural Biotechnology Journal, Volume 21, 2023, Pages 1550-making 1556
- Petersen AB, Tøndervik A, Gaardløs M, Ertesvåg H, Sletta H, Aachmann FL. Mannuronate C-5 Epimerases and Their Use in Alginate Modification. Essays in Biochemistry. 2023;67(3):615-27.
Session 2 – Thursday kl 10.15-11.15

Maria Wilhelmsen Hoff
Maria Wilhelmsen Hoff1, Peik Haugen1, Terje Vasskog1, Njål Rauø2, Lindsey Martinsen3, Johann Eksteen4 & Gøril Laugsand5
(1) UiT Norges arktiske universitet, (2) PHARMAQ, (3) Amicoat, (4) Ard Innovation and (5) Alliance Healthcare
Sustainable technological solutions that maximize the utilization of renewable biological resources are essential to the development of the bioeconomy1,2. The key principle of the bioeconomy is the use of natural resources and recycling of these resources. “Green microbiology” is presented as a solution to mitigate the detrimental environmental effects seen across several large industries. These industries include food and energy production, as well as waste management. Microorganisms not only requires less amounts of natural resources for production but are easy to dispose of. A main challenge in the development of sustainable microbial processes is the cost of upscaling3.
By combining the use of a sequential batch reactor (SBR), mainly used in wastewater treatment, with a mixed microbial community (MMC), operational costs are substantially reduced. This is largely due to removing the need for sterile conditions4. In addition, MMCs has the potential to utilize complex feedstocks due to the metabolic diversity. This allows for further reduction of operational costs and has the potential to increase the value of underutilized materials5,6.
Large quantities of organic side streams with low value are produced in the aquaculture industry4. Simultaneously the demand for seafood increases, and with it the need for sustainable feedstocks which ensure both fish welfare and sufficient nutritional quality7.
A challenge with MMCs is the often lengthy selection time. In this project, a feast and famine regime with uncoupled carbon and nitrogen feeding was implemented for internal and external selective pressure4,5. The aim is to establish a stable SBR operation enriched in lipid storing bacteria for production of a bacterial meal with a lipid composition suitable as a fish feed additive.
References:
- European Commission (2020). Bioeconomy. Bioeconomy | Research and Innovation (europa.eu)
- Forskningsrådet, Innovasjon Norge og Siva (2019). Bioøkonomi – felles handlingsplan for forskning og innovasjon | Norges Forskingsråd (www.forskingsradet.no/publikasjoner)
- A. A. Akinsemolu 2023, Environmental Advances 14:100440.
- Marreios et al. 2023, Journal of Environmental Chemical Engineering, 11:110100.
- Oliveira et al. 2017, New Biotechnology, 37(A), 69-79.
- De Groof et al. 2019, Molecules, 24(3):398.
- Sprague et al. 2016, 2006–2015. Scientific Reports 6:21892.

Mary Dayne Sia Tai
Mary Dayne Sia Tai1, Marte Innselset Flydal2, Gloria Gamiz-Arco1, Trond-André Kråkenes1, Christer Flagtvedt
Didriksen1, Juha Pekka Kallio1 & Aurora Martinez1
(1) Department of Biomedicine, University of Bergen, Bergen; (2) Department of Medical Genetics, Haukeland University Hospital
Hyperphenylalaninemia (HPA) primarily results from pathogenic variants in the PAH gene, which encodes phenylalanine hydroxylase (PAH), the enzyme responsible for converting L-Phe to L-Tyr. Variants of DNAJC12 also cause hyperphenylalaninemia along with dystonia, intellectual disability and neurotransmitter deficiencies in patients without any variants in PAH, in other tetrahydrobiopterin (BH4) dependent hydroxylases or in enzymes involved in BH4 synthesis or regeneration. As an Hsp40 protein, DNAJC12 binds to its client proteins, such as PAH, and presents them to Hsp70 for proper protein folding and homeostasis. However, the mechanism by which DNAJC12 binds to PAH is currently unknown. Human DNAJC12 and PAH, wild-type (WT) and HPA-associated variants, were recombinantly expressed in E. coli and purified before in vitro complex reconstitution. Biophysical and biochemical methods such as analytical size exclusion chromatography (SEC), native PAGE, immunoblotting and dynamic light scattering (DLS) were used to confirm complex formation and investigate the effect of complex formation on the stability of PAH. DNAJC12 and PAH form a complex that can be purified for further characterization. Results from SDS-PAGE, native PAGE and immunoblotting confirm the co-migration of DNAJC12 and PAH in non-denaturing conditions. By monitoring the time-dependent self-aggregation of PAH and HPA associated variants over time using DLS, DNAJC12 was also found to significantly delay PAH aggregation in vitro. Removal of an evolutionarily conserved octapeptide sequence in DNAJC12 was found to abolish its ability to bind to PAH, indicating the significance of this motif for DNAJC12 client binding. DNAJC12 recognizes and binds PAH through an evolutionarily-conserved octapeptide sequence. The binding of DNAJC12 stabilizes PAH, preventing its self-aggregation over time.

Mina Gravdahl
Mina Gravdahl1, Olav A. Aarstad1, Agnes B. Petersen1, Stina G. Karlsen1, Ivan Donati2, Mirjam Czjzek3, Ove Alexander Høgmoen Åstrand4, Philip D. Rye4, Anne Tøndervik5, Håvard Sletta5, Finn L. Aachmann1 & Gudmund Skjåk-Bræk1
(1) Norwegian Biopolymer Laboratory, Department of Biotechnology and Food Science, Norwegian University of Science and Technology, Trondheim, Norway, (2) Department of Life Sciences, University of Trieste, Trieste, Italy, (3) Statiotation Biologique de Roscoff (SBR), Sorbonne Université, Roscoff, France, (4) AlgiPharma AS, Sandvika, Norway, (5) Department of Biotechnology and Nanomedicine, SINTEF Industry, Trondheim, Norway.
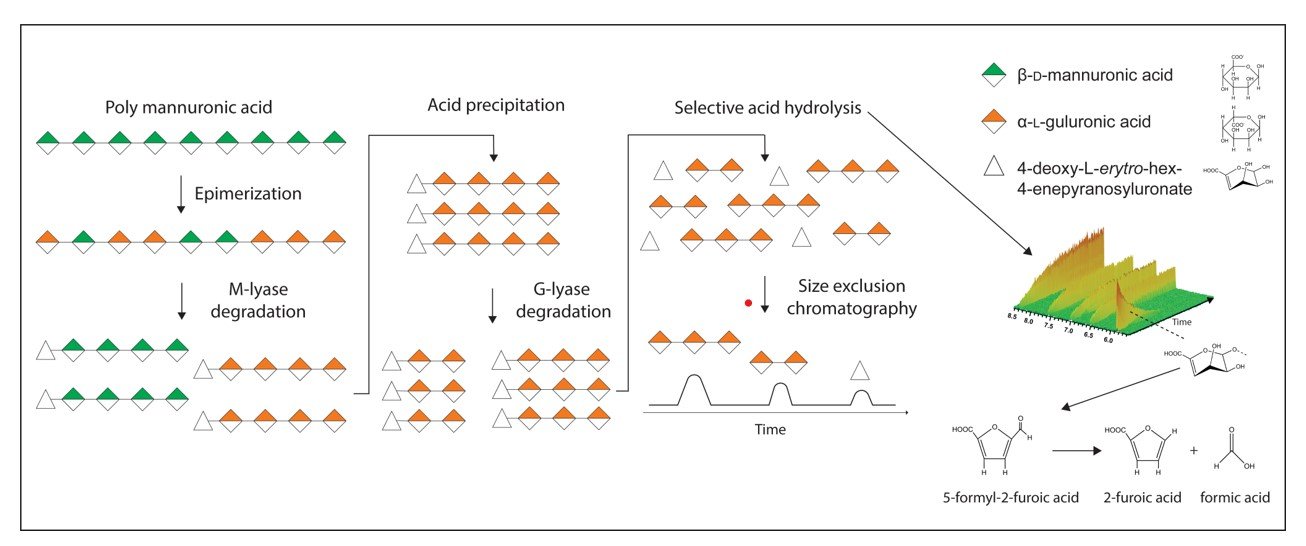
Oligosaccharides from uronic acid-containing polysaccharides can be produced either by chemical or enzymatic degradation. The benefit of using enzymes, called lyases, is their high specificity for various glycosidic linkages. Lyases cleave the polysaccharide chain by an β-elimination reaction, yielding oligosaccharides with an unsaturated sugar (4-deoxy-L-erythro-hex-4-enepyranosyluronate) at the non-reducing end. In this work we have systematically studied acid degradation of unsaturated uronic acid oligosaccharides. Based on these findings, a method for preparing saturated oligosaccharides by enzymatic degradation of uronic acid-containing polysaccharides was developed. This results in oligosaccharides with a pre-defined distribution and proportion of sugar residues compared to the products of chemical degradation, while maintaining the chemical structure of the non-reducing end. The described method was demonstrated for generating saturated oligosaccharides of alginate, heparin and polygalacturonic acid. In the case of alginate, the ratio of hydrolysis rate of Δ-G and Δ-M linkages to that of G-G and M-M linkages, respectively, was found to be approximately 65 and 43, at pH* 3.4, 90 ˚C. Finally, this method has been demonstrated to be superior in the production of α-L-guluronate oligosaccharides with a lower content of β-D-mannuronate residues compared to what can be achieved using chemical depolymerization alone.

Robin Jeske
Robin Jeske & Richard A. Engh
Department of Chemistry (NORSTRUCT), UiT – The Arctic University of Norway, Tromsø
The thermodynamics and kinetics of ligand (L) binding to enzymes (E) in biophysical studies and drug design applications is typically characterized assuming a simple association reaction scheme:
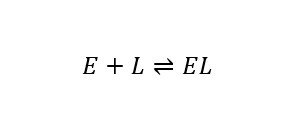
which is associated with a Gibbs energy of binding (ΔGbind), an equilibrium constant, and on and off rates (kon, koff) of binding. This may be misleading in a biological context, as it obscures important physical properties of the binding process, including especially structural multiplicity or disorder and the diversity of chemical environments in molecularly crowded spaces. A more complete scheme may be abbreviated as:
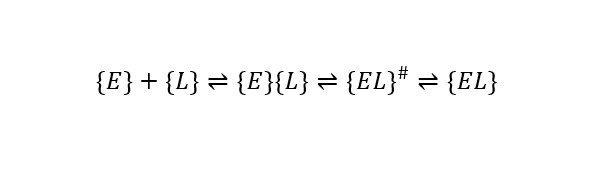
Here, the brackets indicate sets of differing structures whose statistical distributions depend on the changing biological environment, including the proximity of the binding partner. The “#” sign marks a set of higher energy transition state type complexes, and the lower energy final bound state may also be a set of structural conformations. Binding involves a trajectory across a total energy landscape that encodes the process into a reciprocal dance of the binding partners (with their individual average monomeric energy landscapes modulated by proximities of each other, and of other species on the molecularly crowded “dance floor” of the biological environment).
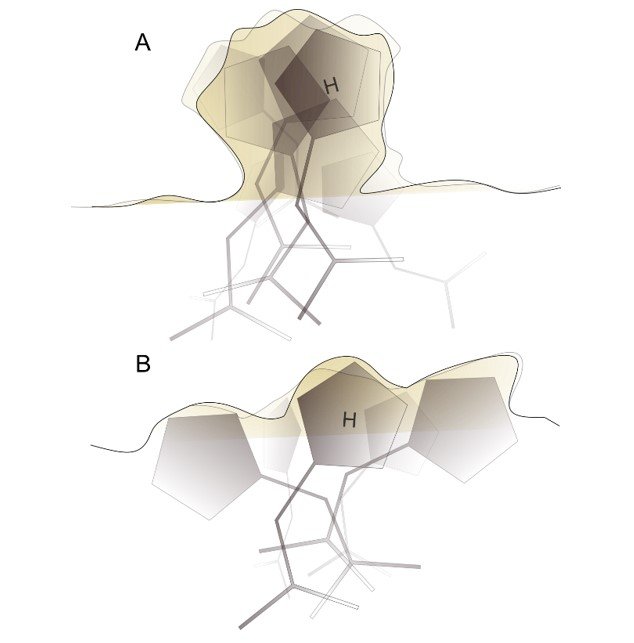
The disorder corresponds to the entropy of the specific averaged states along the reaction coordinate. The figure at right illustrates the entropies of rapidly interconverting structures of a single state (A) and of more distinct structural states, likely with slower interconversion rates (B), and likely with lowered enthalpy. Detailed structural and biophysical studies are needed to characterize entropy across the reaction coordinate.
This has practical implications for peptidic drug design, important for our efforts to design peptide ligands of DYRK1A and other protein kinases. Because peptides are highly flexible compared to typical small molecules, entropy considerations are especially important. Prioritizing compounds that modelling programs predict to have the best binding enthalpies may not be the best approach. We hypothesize that compounds predicted to have multiple good binding poses should be given special weight. Such compounds have lower entropic penalties of binding, but would also have potentially faster on-rates and also would be less susceptible to errors introduced by the simulation method.
Session 3 – Thursday kl 15.15-16.15

Susanne Hansen Troøyen
Susanne Hansen Troøyen, Davide Luciano, & Gaston Courtade
Department of Biotechnology and Food Science, Norwegian University of Science and Technology (NTNU). Trondheim, Norway
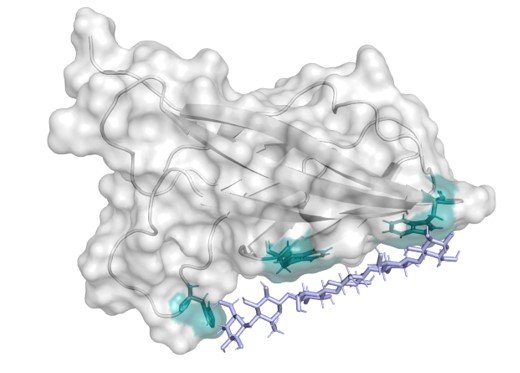
Several carbohydrate-active enzymes contain carbohydrate-binding modules (CBMs) that regulate enzymatic activity by localizing the catalytic domain towards the surface of insoluble substrates such as cellulose [1]. Recently, some CBMs have also been shown to have affinity for non-natural substrates such as polyethylene terephthalate (PET) [2, 3]. CBMs are thought to act as anchors on the substrate surface, allowing the enzyme to perform its activity inside a radius limited by the length of the flexible linker connecting the two domains. However, as illustrated by the Sabatier principle, the CBM should not stay attached at the same position too long – otherwise it would limit the catalytic efficiency of the enzyme. The dynamics of exchange between the free and bound state of the binding module is thus an important, but largely unexplored property of these proteins. NMR spectroscopy offers an opportunity to study protein exchange processes through carefully chosen experiments such as dark state exchange saturation transfer (DEST), solvent paramagnetic relaxation enhancement (sPRE) and relaxation rate measurements. We present here our ongoing investigation into CBM binding dynamics and identification of their substrate-binding site. In combination with affinity assays and kinetic experiments, we anticipate that these insights will contribute to developing our understanding of CBM binding mechanisms.
References:
- Courtade, G., Forsberg, Z., Heggset, E. B., et al. (2018) The carbohydrate-binding module and linker of a modular lytic polysaccharide monooxygenase promote localized cellulose oxidation. J Biol Chem 293(34), 13006 –13015.
- Weber, J., Petrović, D., Strodel, B. et al. (2019) Interaction of carbohydrate-binding modules with poly(ethylene terephthalate). Appl Microbiol Biotechnol 103, 4801–4812.
- Rennison, A. P., Westh, P., and Møller, M. S. (2023) Protein-plastic interactions: The driving forces behind the high affinity of a carbohydrate-binding module for polyethylene terephthalate. Sci Total Environ 870, 161948.

Szymon Mikolaj Szostak
Szymon Szostak & Reidar Lund
Department of Chemistry, University of Oslo, Norway
Peptoids (N-substituted polyglycines) are a relatively new group of synthetic polymers, synthesized for the first time in 1992,[1,2] designed to mimic peptides, while bearing some advantages like enzymatic stability or higher side chain diversity.[3] Differently from peptides, in peptoids the side chain is attached to the nitrogen atom instead of the α-carbon position in the polyglycine backbone. That small change results in the absence of backbone chirality and internal hydrogen bonding, whereas tertiary structure depends entirely on side groups’ interactions.[4]
In this work, we present a new class of monodisperse poly(ethylene glycol)45-peptoid (mPEG45-peptoid) conjugates that self-assemble into stable micelles with amorphous inner core, crystalline outer core, and diffuse PEG shell. We employed Small Angle X-ray Scattering (SAXS) with model analysis, Matrix-Assisted Laser Desorption/Ionization – Mass Spectrometry (MALDI-MS), Pendant Drop Tensiometry (PDT), and Differential Scanning Calorimetry (DSC) for detailed characterization of the compounds and self-assembled structure in biologically relevant pH range. The results give important insight into the process of sequence-dependent peptoid self-assembly showing the possibility of multi-layered nanoparticles which can be potentially used for an adjustable encapsulation system, in which the composition of the core can be altered to interact most efficiently with a desired molecule. The outer crystalline shell provides additional thermodynamic stability and an additional protective layer for the cargo and environment.
References:
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg, C. K. Marlowe, Proc. Natl. Acad. Sci. 1992, 89, 9367–9371.
- R. N. Zuckermann, Pept. Sci. 2011, 96, 545–555.
- A. Battigelli, Biopolymers 2019, 110, e23265.
- N. Gangloff, J. Ulbricht, T. Lorson, H. Schlaad, R. Luxenhofer, Chem. Rev. 2015, 116, 1753–1802.

Tobias Rindfleisch
Tobias Rindfleisch1, Jarl Underhaug2, Hanne Antila3 & Markus Miettinen4
(1) Computational Biology Unit and Department of Chemistry, University of Bergen, (2) Department of Chemistry, University of Bergen, (3) Department of Biomedicine, University of Bergen, (4) Computational Biology Unit and Department of Chemistry, University of Bergen
Keywords: Molecular Microscope, MD simulations, IDPs, NMR relaxation, Force field development
Intrinsically disordered proteins (IDPs) are defined by a lack of a specific 3D structure in aqueous solution. They behave like flexible and strongly dynamic random coils, but demonstrate a characteristic distribution over the conformational space, the so-called structural ensemble. However, IDPs perform important and specific functions at the cellular level and at the super cellular level of the organism.
Molecular dynamics (MD) simulations of IDPs represent a complex problem, because none of the common IDP-specific force fields – a model in MD which defines and parameterizes the interactions of atoms – are able to describe the dynamics and thus the flexible motions of disordered proteins accurately. In consequence, the development of a force field, which is capable of reproducing the dynamics for IDPs correctly, as confirmed by validation against experimental NMR data, is of special importance.
The key-idea behind this strategy is to combine the accuracy and precision of NMR experiments with the highly intuitive visualization of MD simulations, such that NMR data can be finally interpreted via the full-atomistic representation of MD – which ultimately describes the creation of a molecular microscope.
This project is motivated by the findings of Oh et al. (2012) that dipeptides behave similarly to disordered proteins, suggesting that these elementary building blocks can be used to test and, if needed, calibrate an IDP-specific MD model.
A gradient-free evolutionary approach will be used to automatically optimize a previously selected and well performing force field; here the force field parameters are iteratively adjusted to obtain an improved match between the resulting protein dynamics in MD simulations and in NMR experiments (relaxation times, homo and hetero nuclear Nuclear Overhauser Effect). Importantly, the corresponding NMR observables can be computed from the MD simulations directly, without assuming an intervening model. The NMR experiments are performed at the Norwegian NMR Platform.

Zuzanna Justyna Samol
Zuzanna Samol1, Magne O. Sydnes2, & Erik Agner3
(1) Polypure AS, Norway, Department of Chemistry, Bioscience and Environmental Engineering, University of Stavanger, Norway, (2) Department of Chemistry, University of Bergen, Norway, (3) Polypure AS, Norway
Capsaicin, a well-known spicy compound found in chili peppers, binds selectively to one of the vanilloid receptors, TRPV1 (transient receptor potential vanilloid 1). When capsaicin is delivered in a high enough dose to the receptor, it can provide pain relief. Since these receptors are prevalent in sensory neurons, they are an interesting target for the treatment of chronic neuropathic pain.1
Capsaicin is often applied topically through creams and patches. However, these medications have several drawbacks. Creams deliver low quantities of the compound, requiring multiple reapplications throughout the day. High-concentration patches, due to their pungency and skin-burning effects, must be applied with a local anesthetic. This results in limited efficacy and patient compliance.2,3 Therefore, there is a need to develop topical formulations with enhanced drug loading capacity and minimized side effects.
Our work focused on improving the properties of the polymeric excipients used in hydrogels for drug delivery. The employed strategies included the synthesis of monodisperse PEG and PPG derivatives, which provided defined thermosensitive copolymers. We have also investigated the effect of incorporation high-purity PPG-8 oligomer into the hydrogel. This enhanced the gelation characteristics, as well as in vitro release of capsaicin. In combination, these approaches offer higher purity and reproducibility of the hydrogel matrix, as well as higher hydrophobic payloads with improved delivery.
References:
- Yang F, Zheng J. Understand spiciness: mechanism of TRPV1 channel activation by capsaicin. Protein Cell. 2017;8(3):169-177. doi:10.1007/s13238-016-0353-7
- Avila F, Torres-Guzman R, Maita K, et al. A Review on the Management of Peripheral Neuropathic Pain Following Breast Cancer. Breast Cancer (Dove Med Press). 2023;15:761-772. doi:10.2147/BCTT.S386803
- Thouaye M, Yalcin I. Neuropathic pain: From actual pharmacological treatments to new therapeutic horizons. Pharmacol Ther. 2023;251:108546. doi:10.1016/j.pharmthera.2023.108546