
3 Minute Flash Presentations 2024
Wednesday – Session 1

Davide Luciano
Davide Luciano1, Jochen Schmid2, & Gaston Courtade1
(1) Department of Biotechnology and Food Science, NTNU Norwegian University of Science and Technology, Trondheim, Norway, (2) Institute of Molecular Microbiology and Biotechnology, University of Münster, Münster, Germany
Keywords: Glycosyltransferase, GTB, Xanthan Gum, biased MD
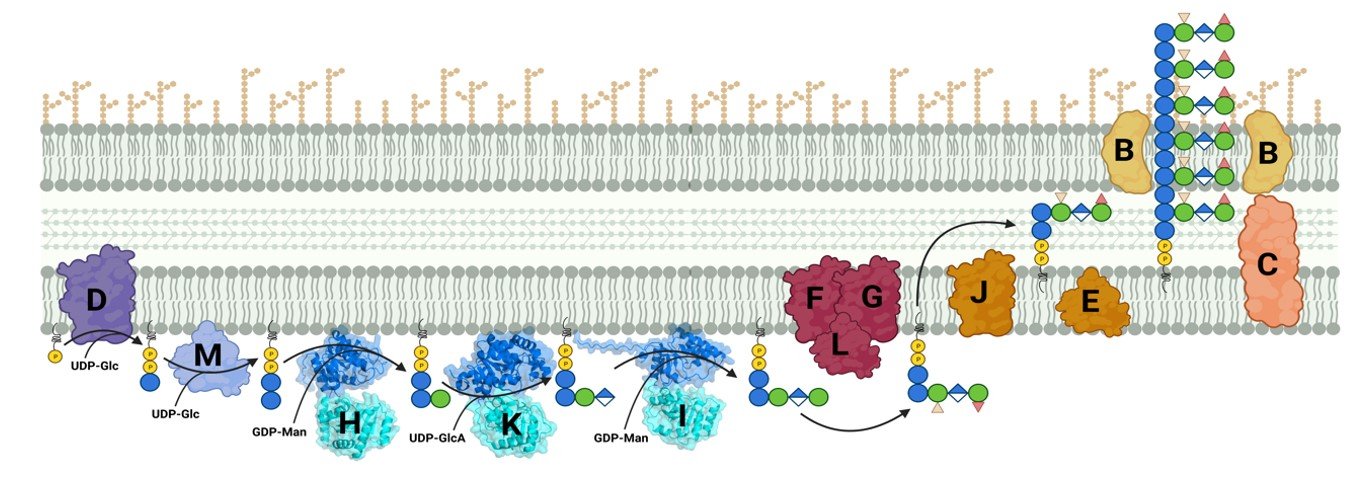
Exopolysaccharides are a diverse class of molecules with a wide range of industrial applications. Xanthan gum, a heteropolysaccharide synthesized by the Gram-negative bacterium Xanthomonas campestris,[1] is a significant contributor to this class of polymers. Its versatile mechanical properties make it useful in various fields such as the food, material, and pharmaceutical industries.[2] Modifications of the polysaccharide are being studied to modulate its properties for more specific applications.[3] In this study, we present a computational approach to identify key residues in the active site of GumK, a glycosyltransferase involved in xanthan biosynthesis, that influence the dynamics and activity of the enzyme. We describe the dynamics of the two domains and the residues involved in the binding of the natural substrates. Our computational protocol is applicable to other glycosyltransferases within the pathway and can elucidate enzyme selectivity. This insight can inform bioengineering strategies to produce new variants of xanthan gum.
References:
- Anke Becker and Federico Katzen and Alfred Pühler and Luis Ielpi (1998) Xanthan gum biosynthesis and application: a biochemical /genetic perspective. Applied Microbiology and Biotechnology, 50: 145-152
- Chaturvedi Surabhi, Kulshrestha Sanchita, Bhardwaj Khushboo and Jangir Rekha (2021) A Review on Properties and Applications of Xanthan Gum. Microbial Polymers: Applications and Ecological Perspectives: 87-107
- Patel, Jwala and Maji, Biswajit and Moorthy, N. S. Hari Narayana and Maiti, Sabyasachi (2020) Xanthan gum derivatives: review of synthesis, properties and diverse applications. RSC Adv, 10(45): 27103-27136

Eva Smorodina
Eva Smorodina1, Oliver Crook2, Rahmad Akbar1, Puneet Rawat1, Dario Segura Pena3, Nikolina Sekulic3, Ole Magnus Fløgstad3, Khang Lê Quý1, Brij Bhushan Mehta1, Johannes Loeffler4, Monica Fernandez-Quintero4, Hannah Turner4, Andrew B. Ward4, & Victor Greiff1
(1) Department of Immunology, University of Oslo and Oslo University Hospital, Oslo, Norway, (2) Department of Statistics, University of Oxford, Oxford, (3) Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, Faculty of Medicine, University of Oslo, Oslo, Norway, (4) Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, USA
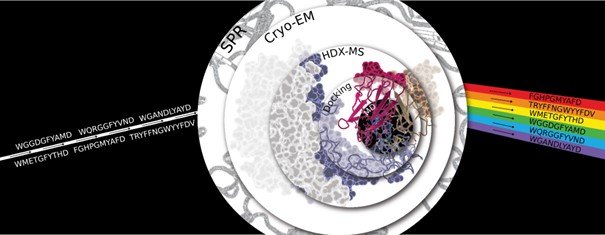
Antibodies are key therapeutics but the principles behind diverse paratopes binding to the same epitope remain unexplained. An insufficient understanding of the structural rules behind antibody-antigen binding, due to a lack of experimentally resolved structures, leads to the current inability to characterize antibody variants binding in silico. Here we propose a rule-based antibody design that relies on a thorough understanding of epitope-paratope interactions, in contrast to generative design based on millions of trials and errors. We identified the epitope of five affinity-verified Trastuzumab variants using cryo-EM and position-resolved HDX-MS. Rigid models alone are insufficient for accurate antibody-antigen modeling while molecular dynamics simulations with computational analysis of the complex conformations succeed in replicating and complimenting experimental findings. Structural parameters calculated based on geometry, surface, and biochemical properties were able to distinguish between high and low binders. We highlight the possibilities of AI in antibody and antibody-antigen structure modeling, demonstrating the limitations of various language-based models to predict and understand antibody variants. Overall, our study explains the binding mechanisms of the variant sequences, showing how antibodies with diverse sequences share similar antigen-binding rules.

Greta Nardini
Greta Nardini1, Kristian Hovde Liland2, Sileshi Gizachew Wubshet3, Nils Kristian Afseth3 & Kenneth Aase Kristoffersen1
(1) NMBU – Norwegian University of Life Science, KBM Faculty, P.O. Box 5003, NO-1432, Ås, Norway, (2) NMBU – Norwegian University of Life Science, REALTEK Faculty, P.O. Box 5003, NO-1433, Ås, Norway, (3) Nofima – Norwegian Institute of Food, Fisheries and Aquaculture Research, P.O. Box 210, NO-1431, Ås, Norway
Enzymatic Protein Hydrolysis (EPH) is a well-established and versatile technology for the valorization of protein from food industry side-streams. Low-value cuts from poultry, a protein-rich biomass, are transformed into high-value protein hydrolysates with different physicochemical properties. One example is collagen-enriched hydrolysates used in food products, pharmaceuticals, and cosmetics.[1]
Process monitoring and feed-forward control mechanisms are crucial to optimize yield and product quality from EPH processes.[2] The amino acid composition is an important process parameter, and along with the molecular weight distribution (MWD), it is a key parameter in determining the hydrolysates’ physicochemical properties. In poultry side-stream valorization, collagen solubilization is another key parameter and can be followed using Hydroxyproline, an amino acid almost exclusive to collagen.[3]
The gold standard for amino acid composition analysis is the chromatographic method.[4] Recently, however, there has been a growing interest in spectroscopic techniques which are green, rapid, and non-destructive, making them ideal for real-time measurements.[2,5,6] Fourier Transform Infrared spectroscopy (FTIR), a vibrational spectroscopy technique, has successfully been applied to monitor industrial processes to assess relevant analytical parameters like MWD and collagen content.[7,8] While vibrational spectroscopy is not suitable for predicting the amino acid composition, Nuclear Magnetic Resonance (NMR) spectroscopy is. NMR offers deeper insights because it is superior to FTIR in probing metabolites and low molecular components, it also provides more reliable quantitative (qNMR) data.[9,10]
In this study, the performance of FTIR and NMR spectroscopy has been compared with classical methods for investigating collagen content in EPH samples from poultry side-streams.
References:
- K. A. Kristoffersen et al., Food Chem 2022, 382, DOI 10.1016/j.foodchem.2022.132201.
- S. G. Wubshet et al., Food Bioproc Tech 2018, 11, 2032–2043.
- R. E. Neuman et al., Journal of Biological Chemistry 1950, 184, 299–306.
- European Commission Regulation, No 152/2009 of 27 January 2009.
- S. Shin et al., Food Chem 2021, 352, DOI 10.1016/j.foodchem.2021.129329.
- M. S. Eissa et al., TrAC 2024, 170, DOI 10.1016/j.trac.2023.117435.
- I. Måge et al., LWT 2021, 152, DOI 10.1016/j.lwt.2021.112339.
- K. A. Kristoffersen et al., Spectrochim Acta A 2023, 301, DOI 10.1016/j.saa.2023.122919.
- T. Tchipilov et al., Methods Protoc 2023, 6, DOI 10.3390/mps6010011.
- T. Riemer et al., Biomacromolecules 2012, 13, 2110–2117.
Thursday – Session 2

José Miguel Godoy Muñoz
José M. Godoy Muñoz1, & Petri Kursula1,2
(1) Department of Biomedicine, University of Bergen, Bergen, Norway, (2) Faculty of Biochemistry and Molecular Medicine & Biocenter Oulu, University of Oulu, Oulu, Finland
Nanobodies, or single-domain antibodies, are emerging as state-of-the-art molecular tools for protein science. Their small size and high antigen specificity, among other characteristics, make them versatile tools to study the structure and function of their target proteins. A testament to this comes with the recent characterisation of two nanobodies targeting the activity-regulated cytoskeleton-associated protein (Arc), a complex regulator of synaptic plasticity.
These two nanobodies, E5 and H11, acted as crystallisation chaperones and promoted the structural characterization of the human Arc N-lobe (Arc-NL) at atomic resolution via X-ray crystallography. Both nanobodies bound the multi-peptide binding site of Arc-NL and inhibited the binding of a high-affinity endogenous peptide, as demonstrated using isothermal titration calorimetry. In addition, sequence homology searches of the nanobodies’ CDR3s revealed new potential binding partners of Arc-NL and helped identify the possible binding site of a well-known Arc binding partner (PICK1).
These results are only a fraction of the vast potential of nanobodies. Currently, nanobodies against other proteins of the nervous system have been developed. Our present focus is to extend the use of nanobodies to immunohistochemistry and explore their potential within interdisciplinary research, putting special emphasis on brain myelination.

Juliana Miranda Tatara
Juliana Miranda Tatara1, Victoria Queiroz2,Klara Stensvåg1, Jônatas Abrahão2, & Gabriel Magno De Almeida1
(1) The Arctic University of Norway, Tromsø, Norway, (2) Universidade Federal de Minas Gerais, Brazil
Giant viruses are part of the nucleocytoplasmic large DNA viruses (NCLDVs) group, and they were first described in 2003 [1], leading to a revolution in microbiology. Recent metagenomic research revealed that giant viruses are ubiquitous [2], and their genome size can reach up to 2.5Mb [3]. Such characteristics lead to a remarkable biotechnology potential coded by their still mysterious genomes. The arctic region, such as the Nordic Sea, have been described as hotspots for finding giant viruses [4]. Herein, we aimed to collect different samples above the arctic circle and use them to isolate giant viruses using Acanthamoeba sp. as hosts, and further explore different compounds produced during infection using one of our isolates as model. So far, we collected over 250 samples around the north of Norway and Nordic Sea, including: marine and freshwater environments, urban, sewage and deep-sea vent samples. All of them were prepared and used for virus isolation by mixing samples and cells, following culturing steps until cytopathic effect (CPE) appeared. Six different samples presented CPE and the presence of a giant virus was confirmed by Transmission Electron Microscopy images. Virus species are still to be confirmed through sequencing analysis, yet morphological features suggest viruses to be part of the marseilleviridae and mimiviridae families. Preliminary results from metabolomics analysis pointed to different compounds being produced during a Norwegian marseillevirus infection in amoeba. Subsequent steps are virus species identification and characterization, and the description of these diverse compounds produced during the infection.
References:
- La Scola, Bernard et al. “A giant virus in amoebae.” Science (New York, N.Y.) vol. 299,5615 (2003): 2033. doi:10.1126/science.1081867
- Schulz, Frederik et al. “Giant virus diversity and host interactions through global metagenomics.” Nature vol. 578,7795 (2020): 432-436. doi:10.1038/s41586-020-1957-x
- Philippe, Nadège et al. “Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes.” Science (New York, N.Y.) vol. 341,6143 (2013): 281-6. doi:10.1126/science.1239181
- Gao, Chen et al. “Viral Characteristics of the Warm Atlantic and Cold Arctic Water Masses in the Nordic Seas.” Applied and environmental microbiology vol. 87,22 (2021): e0116021. doi:10.1128/AEM.01160-2 Abstract text (Calibri 12 point).

Liza Nguyen Van Sang
Liza Nguyen, Emil Lindbäck, & Magne O. Sydnes
UiS
Bacteria have always been a threat to human health, but the hazard and mortality rates have been increased with the emergence of resistance strains.1 Their mechanism of defence and natural evolution have compromised the effectiveness of antibiotics, to the extent that there may be no viable medicine on the market to counter bacterial infection in the future.2
Dual action-based molecules are one of the approaches that can be explored to design new drugs for various diseases, including cancer,3 diabete,4 and alzheimer.5 In the context of bacterial resistance, several hybrid medicines have been synthesized during the past decades, as part of an effort to overcome and find a solution to the global bacterial resistance.6
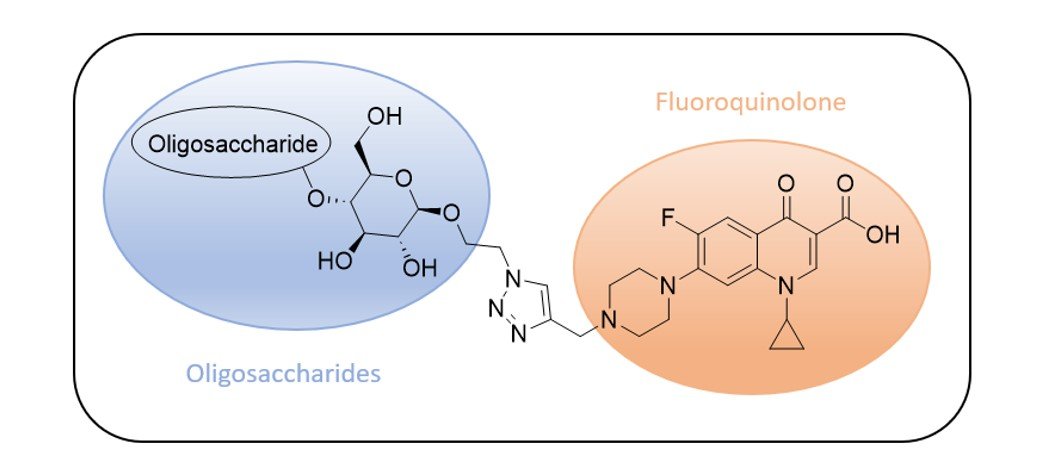
The Marie Curie project “Stop Spread Bad Bugs” (SSBB) aims to develop and test new dual action-based molecules with antibacterial properties. The linkage of a known active scaffold (Ciprofloxacin) with new oligosaccharides offers a combination of their respective pharmaceutical activities and the novelty of the structure is making it more difficult for bacteria to develop simultaneous resistance mechanisms against both moieties
References:
- Baindara, P.; Mandal, S. M. Protein Pept. Lett. 2019, 26, 324-331.
- World Health Organization,Antibacterial agents in clinical development : an analysis of the antibacterial clinical development pipeline, including tuberculosis, 2017.
- Rou, R.; Ria, T.; RoyMahaPatra, D.; Hossain Sk, U. ACS Omega, 2023, 19, 16532-16544.
- Woolston, C., Nature, 2013, 14062.
- Cheong, S.L.; Tiew, J.K.; Fong, Y.H.; Leong, H.W.; Chan, Y.M.; Chan, Z.L.; Kong, E.W.J. Pharmaceuticals, 2022, 15, 1560.
- Bremner, J. B.; Ambrus, J. I.; Samosorn, S. Curr. Med. Chem., 2007, 14, 1459-1477.
- Roemhild, R.; Bollenbach, T.; Andersson, D. I. Nat. Rev. Microbiol. 2022, 20, 478-490.
Thursday – Session 3

Nadia Aftab
Nadia Aftab, Jonathan Hira1, Bhupender Singh1, Johanne U. Ericson1, Mona Johannessen1, & Christian S. Lentz1
Research Group for Host-Microbe Interaction, Department of Medical Biology, UiT – The Arctic University of Norway, Tromsø, Norway
Bacterial pathogens have evolved a variety of adaptation strategies to thrive in ever changing environment (1). Phenotypic heterogeneity, often acts as a short-term adaptive mechanism, enabling an isogenic population to cope better with environmental challenges (2). An example of this is the presence of distinct sub-populations with varying ability to adapt and grow in new milieu where some bacteria can flourish in novel nutritional conditions, while others cannot (3). This variability in growth is a critical aspect of bacterial physiology, playing a vital role in their ability to colonize diverse niches within a host environment. Given this background, we hypothesize that by cultivating isogenic bacteria under different growth conditions, we can identify and quantify the sub-populations that vary in their adaptability. Therefore, we established a simplistic invitro model that led to systematically study and characterize these growth phenotypes in Staphylococcus aureus, an important human pathogen. Our results showed a great deal of variability across different sub-population within a clonal population. Further investigations are underway to identify the molecular factors contributing to these distinct phenotypes and to explore their mechanisms of action, aiming to enhance our understanding of this adaptive phenomenon.
References:
- Martino ME, Joncour P, Leenay R, Gervais H, Shah M, Hughes S, et al. Bacterial Adaptation to the Host’s Diet Is a Key Evolutionary Force Shaping Drosophila-Lactobacillus Symbiosis. Cell Host Microbe. 2018;24(1):109-19.e6.
- Kundu K, Weber N, Griebler C, Elsner M. Phenotypic heterogeneity as key factor for growth and survival under oligotrophic conditions. Environmental Microbiology. 2020;22(8):3339-56.
- Reyes Ruiz LM, Williams CL, Tamayo R. Enhancing bacterial survival through phenotypic heterogeneity. PLoS Pathog. 2020;16(5):e1008439.

Natalia Mojica Cortes
Natalia Mojica1, Flore Kersten1,2, Albert Serrano3, Joel B. Heim1, Gabriele Cordara1, Ken Teter3, & Ute Krengel1
(1) Department of Chemistry, University of Oslo, (2) Centre for Molecular Medicine Norway (NCMM), University of Oslo, (3) Burnett School of Biomedical Sciences, University of Central Florida
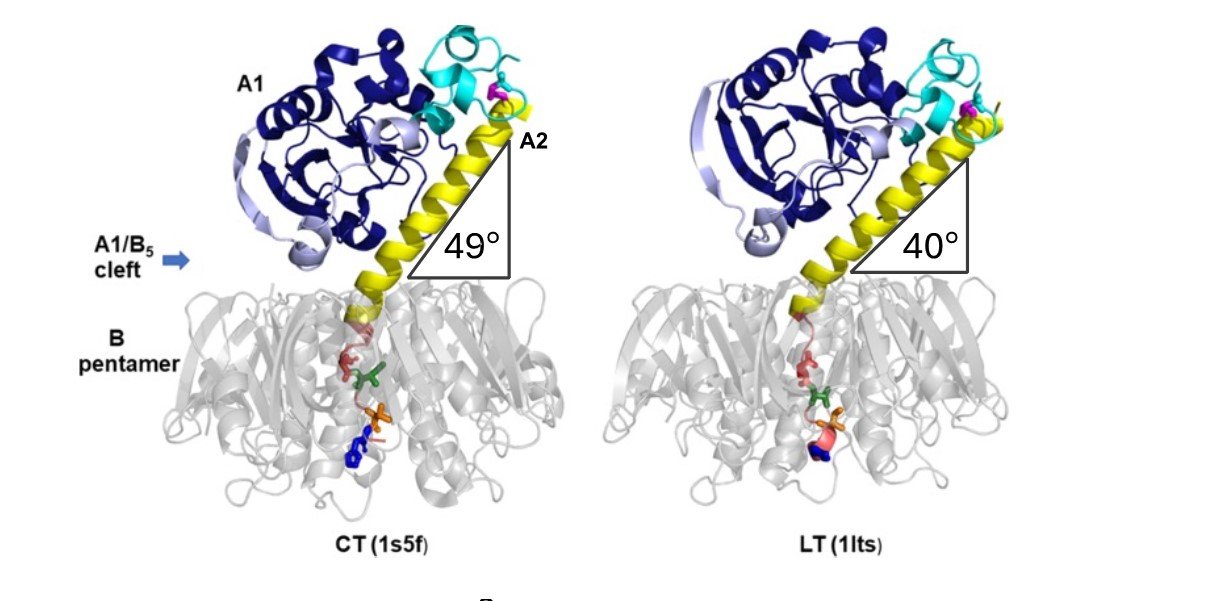
Cholera toxin (CT) and heat-labile enterotoxin (LT) are two similar AB5 toxins responsible for the diarrhea characteristic of Vibrio cholerae and enterotoxigenic Escherichia coli (ETEC) infections. They consist of a catalytically active A1 subunit, an A2 linker, and a pentamer of cell-binding B-subunits 1. Both toxins bind to the same GM1 surface receptor on the host cells and have similar levels of enzymatic activity, yet CT is more potent than LT, making cholera the more severe disease. The difference in toxicity has been attributed to structural differences near the C-terminus of the A2 linker (amino acid residues 226-236) 2, but the underlying molecular mechanism remains unknown. Recently, we showed that toxin disassembly by protein disulfide isomerase (PDI), which is a key event in the intoxication process, is more efficient for CT than for LT3. We hypothesized that the difference in toxin disassembly is related to the positioning of the A1 subunit relative to the B-pentamer3 (Figure 1).
Here, we determined the crystal structures of two cholera toxin variants where either one (D229E) or four (D229E, I230V, T232I, H233Y) amino acid residues in the critical A2 linker sequence were substituted for the residues present in LTA2 (Figure 1; colored residues within the pores of the grey pentamers). The results of this structural analysis will be presented here.
References:
- Heggelund, J. E., Bjørnestad, V. A., and Krengel, U. (2015) Vibrio cholerae and Escherichia coli heat-labile enterotoxins and beyond, in The Comprehensive Sourcebook of Bacterial Protein Toxins, pp 195–229. Elsevier Ltd.
- Rodighiero, C., Aman, A. T., Kenny, M. J., Moss, J., Lencer, W. I., and Hirst, T. R. (1999) Structural Basis for the Differential Toxicity of Cholera Toxin and Escherichia coli Heat-labile Enterotoxin. Journal of Biological Chemistry 274, 3962–3969.
- Serrano, A., Guyette, J. L., Heim, J. B., Taylor, M., Cherubin, P., Krengel, U., Teter, K., and Tatulian, S. A. (2022) Holotoxin disassembly by protein disulfide isomerase is less efficient for Escherichia coli heat-labile enterotoxin than cholera toxin. Sci Rep 12, 34.

Ida Marie Stephansen
Ida Marie Stephansen1 & Fernando Pérez-García1*
(1) Department of Biotechnology and Food Science, Norwegian University of Science and Technology, 7491 Trondheim, Norway
This project aims to meet the increasing demand for sustainable resources driven by a growing population. New renewable and alternative feedstocks are continuously sought in microbial biotechnology, and seaweed presents a promising solution. In particular, we aim to engineer the bacterium Corynebacterium glutamicum, which is commonly used as an industrial workhorse for the large-scale production of amino acids[1]. By applying molecular and synthetic biology tools, we will enable the utilization of seaweed-derived sugars like mannitol, xylose, or galactose by C. glutamicum strains[2]. Our approach will focus on co-cultivation, where each C. glutamicum strain will be engineered to utilize a specific sugar. We hypothesize that this method may improve the carbon conversion yield compared to classic monoculture approaches[3]. To support the concept of circular bioeconomy, utilization of seaweed sugars will be coupled with the production of added-value compounds like amino acids. Finally, to prove the potential of this idea, the newly established co-cultures will be scaled up in bioreactors using red and brown seaweed hydrolysates as the carbon source. Hence, this research seeks to contribute to sustainable production by tapping into the vast potential of ocean resources.
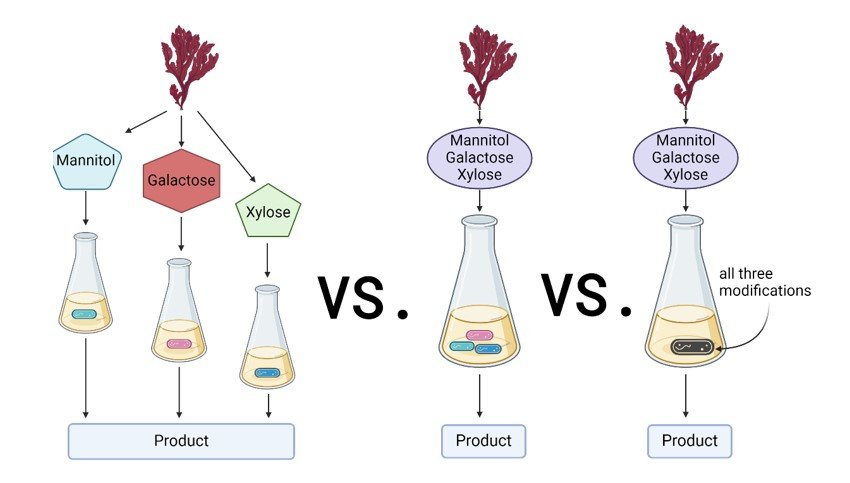
References:
- Wolf, Sabrina, et al. “Advances in metabolic engineering of Corynebacterium glutamicum to produce high-value active ingredients for food, feed, human health, and well-being.” Essays in biochemistry 65.2 (2021): 197-212.
- Zhong, Haowei, et al. “The structural characteristics of seaweed polysaccharides and their application in gel drug delivery systems.” Marine drugs 18.12 (2020): 658.
- Pérez-García, Fernando, et al. “Dynamic co-cultivation process of Corynebacterium glutamicum strains for the fermentative production of riboflavin.” Fermentation 7.1 (2021): 11.